
Coexistence of Myelin Oligodendrocyte Glycoprotein Immunoglobulin G and Neuronal or Glial Antibodies in the Central Nervous System: A Systematic Review


Abstract
:1. Introduction
2. Methods
2.1. Search Strategy and Study Selection
2.2. Data Extraction
2.3. Statistical Analysis
3. Results
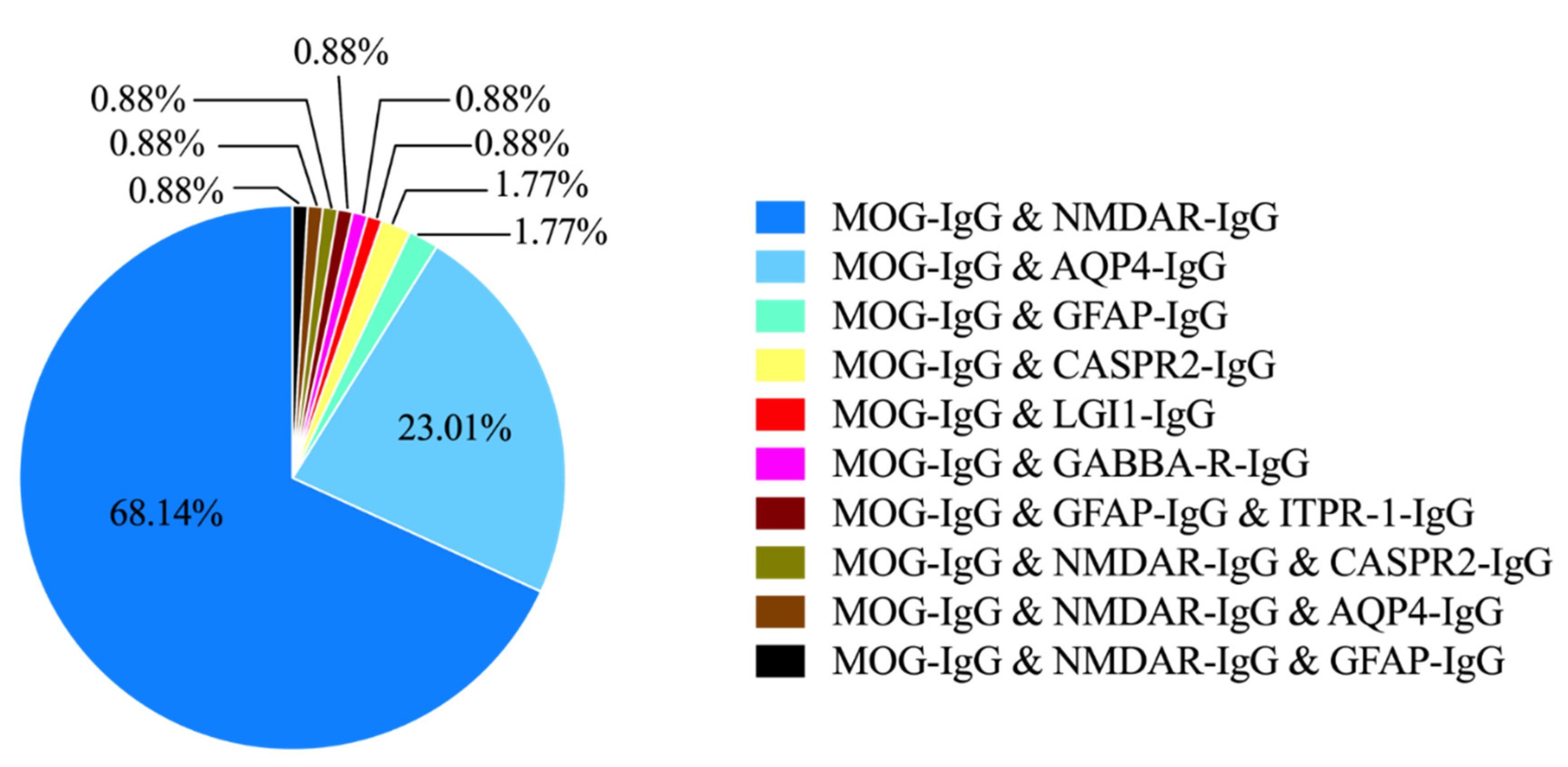
3.1. Study Characteristics and the Spectrum of Coexistence Syndromes
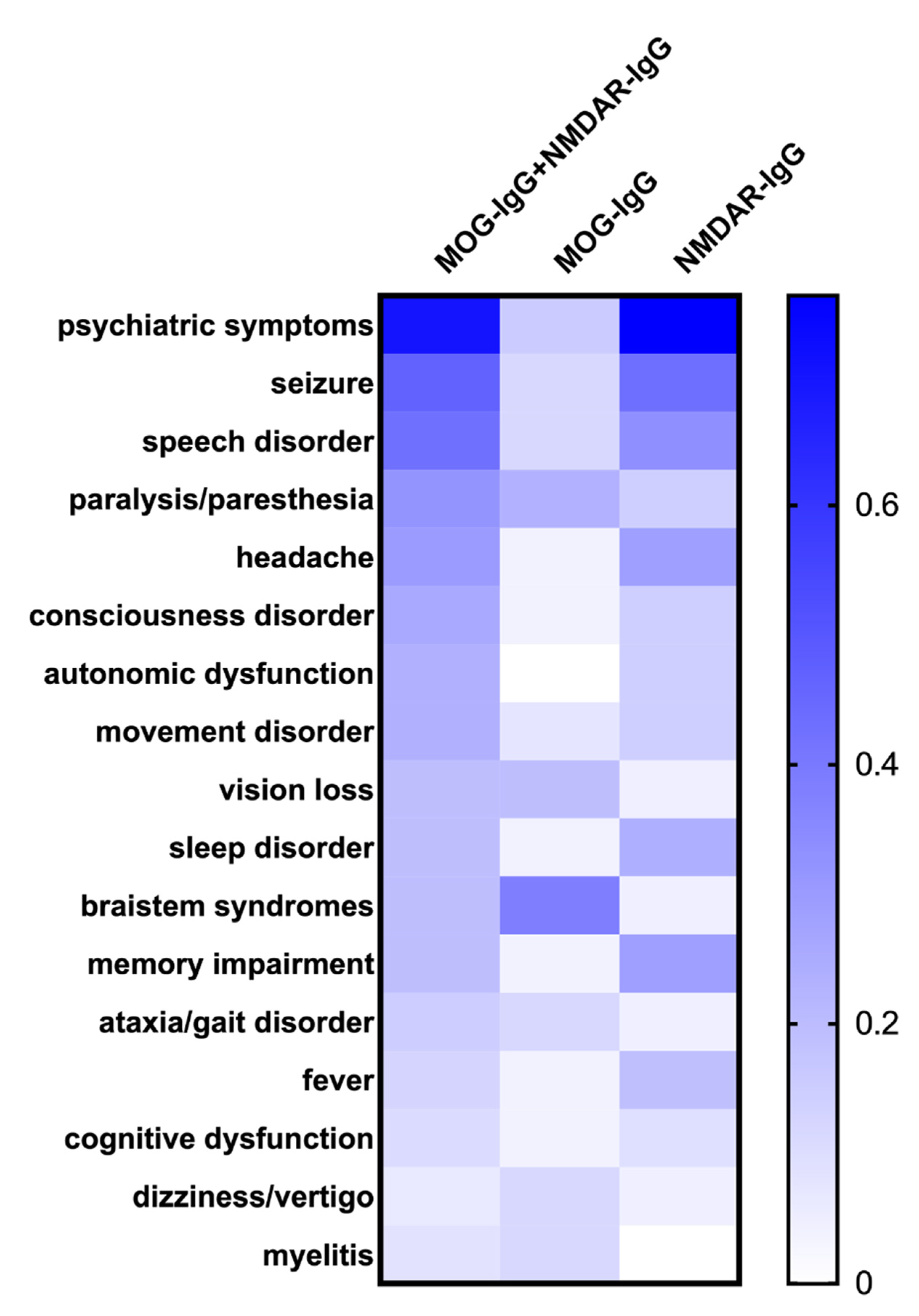
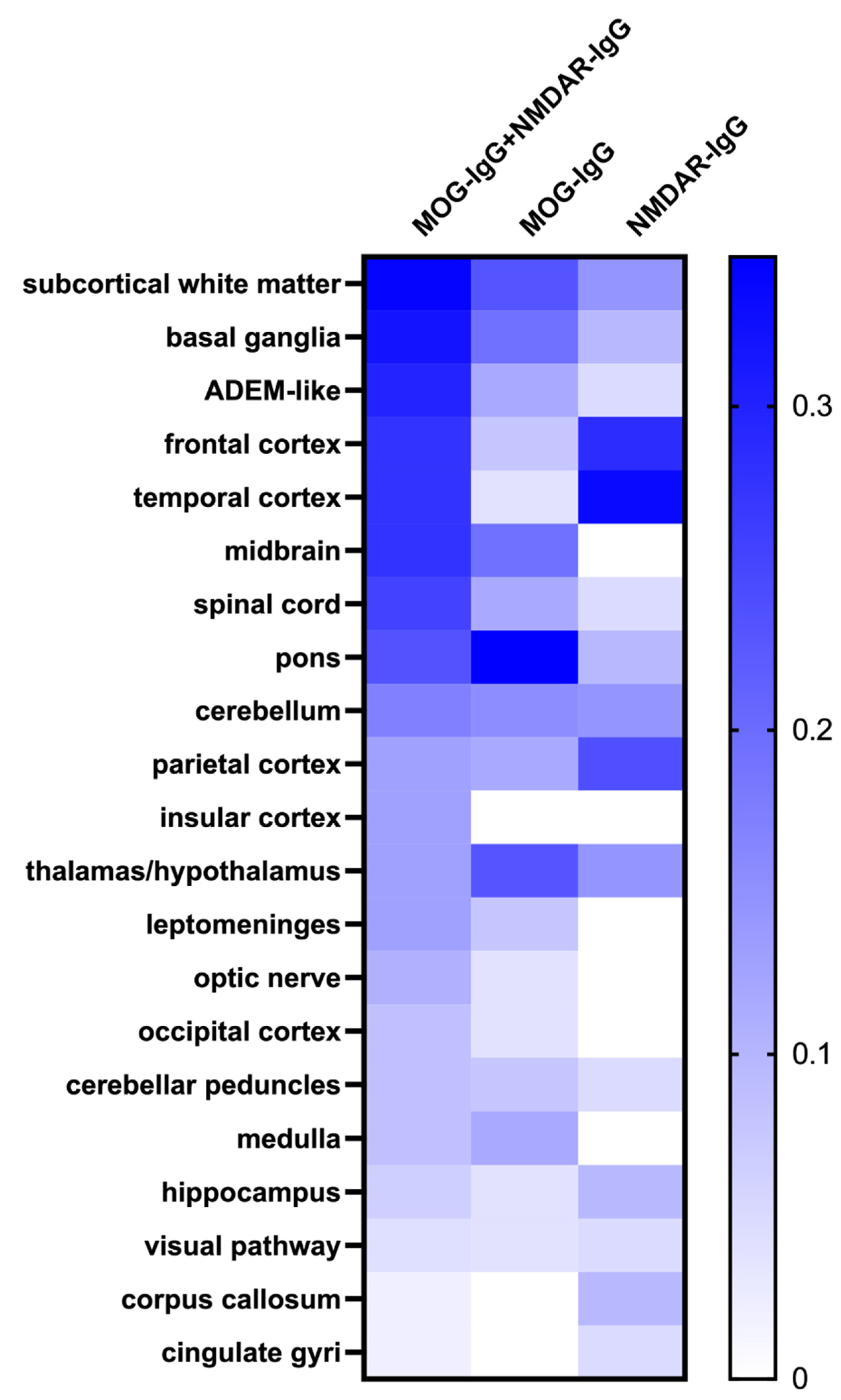
3.2. Coexistence of MOG-IgG and NMDAR-IgG
3.3. Coexistence of MOG-IgG and AQP4-IgG
3.4. Coexistence of MOG-IgG and Other Neuronal or Glial Antibodies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johns, T.G.; Bernard, C.C.A. The Structure and Function of Myelin Oligodendrocyte Glycoprotein. J. Neurochem. 1999, 72, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Marta, C.B.; Montano, M.B.; Taylor, C.M.; Taylor, A.L.; Bansal, R.; Pfeiffer, S.E. Signaling Cascades Activated upon Antibody Cross-Linking of Myelin Oligodendrocyte Glycoprotein. J. Biol. Chem. 2005, 280, 8985–8993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, A.; Bauer, J.; Litzenburger, T.; Schubart, A.; Linington, C. T- and B-Cell Responses to Myelin Oligodendrocyte Glycoprotein in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Glia 2001, 36, 220–234. [Google Scholar] [CrossRef]
- Peschl, P.; Bradl, M.; Höftberger, R.; Berger, T.; Reindl, M. Myelin Oligodendrocyte Glycoprotein: Deciphering a Target in Inflammatory Demyelinating Diseases. Front. Immunol. 2017, 8, 529. [Google Scholar] [CrossRef] [PubMed]
- Menge, T.; Lalive, P.H.; von Büdingen, H.-C.; Genain, C.P. Conformational Epitopes of Myelin Oligodendrocyte Glycoprotein Are Targets of Potentially Pathogenic Antibody Responses in Multiple Sclerosis. J. Neuroinflamm. 2011, 8, 161. [Google Scholar] [CrossRef]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.-A.; Hümmert, M.W.; et al. MOG-IgG in NMO and Related Disorders: A Multicenter Study of 50 Patients. Part 1: Frequency, Syndrome Specificity, Influence of Disease Activity, Long-Term Course, Association with AQP4-IgG, and Origin. J. Neuroinflamm. 2016, 13, 279. [Google Scholar] [CrossRef] [Green Version]
- Kitley, J.; Woodhall, M.; Waters, P.; Leite, M.I.; Devenney, E.; Craig, J.; Palace, J.; Vincent, A. Myelin-Oligodendrocyte Glycoprotein Antibodies in Adults with a Neuromyelitis Optica Phenotype. Neurology 2012, 79, 1273–1277. [Google Scholar] [CrossRef]
- López-Chiriboga, A.S.; Majed, M.; Fryer, J.; Dubey, D.; McKeon, A.; Flanagan, E.P.; Jitprapaikulsan, J.; Kothapalli, N.; Tillema, J.-M.; Chen, J.; et al. Association of MOG-IgG Serostatus with Relapse after Acute Disseminated Encephalomyelitis and Proposed Diagnostic Criteria for MOG-IgG–Associated Disorders. JAMA Neurol. 2018, 75, 1355. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Aktas, O.; Asgari, N.; Dale, R.C.; de Seze, J.; Franciotta, D.; Fujihara, K.; Jacob, A.; Kim, H.J.; et al. MOG Encephalomyelitis: International Recommendations on Diagnosis and Antibody Testing. J. Neuroinflamm. 2018, 15, 134. [Google Scholar] [CrossRef]
- Denève, M.; Biotti, D.; Patsoura, S.; Ferrier, M.; Meluchova, Z.; Mahieu, L.; Heran, F.; Vignal, C.; Deschamps, R.; Gout, O.; et al. MRI Features of Demyelinating Disease Associated with Anti-MOG Antibodies in Adults. J. Neuroradiol. 2019, 46, 312–318. [Google Scholar] [CrossRef]
- Kunchok, A.; Flanagan, E.P.; Krecke, K.N.; Chen, J.J.; Caceres, J.A.; Dominick, J.; Ferguson, I.; Kinkel, R.; Probasco, J.C.; Ruvalcaba, M.; et al. MOG-IgG1 and Co-Existence of Neuronal Autoantibodies. Mult. Scler. J. 2021, 27, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Ren, K.; Wu, J.; Li, H.; Sun, T.; Yan, Y.; Guo, J. Overlapping Syndrome of MOG-IgG-Associated Disease and Autoimmune GFAP Astrocytopathy. J. Neurol. 2020, 267, 2589–2593. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Bai, M.; Yan, X.; Ren, K.; Ding, J.; Zhao, D.; Li, H.; Yan, Y.; Guo, J. Possible Coexistence of MOG-IgG-Associated Disease and Anti-Caspr2 Antibody-Associated Autoimmune Encephalitis: A First Case Report. Ther. Adv. Neurol. Disord. 2020, 13, 175628642096946. [Google Scholar] [CrossRef]
- Ding, J.; Li, X.; Tian, Z. Clinical Features of Coexisting Anti-NMDAR and MOG Antibody-Associated Encephalitis: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 711376. [Google Scholar] [CrossRef]
- Kunchok, A.; Chen, J.J.; McKeon, A.; Mills, J.R.; Flanagan, E.P.; Pittock, S.J. Coexistence of Myelin Oligodendrocyte Glycoprotein and Aquaporin-4 Antibodies in Adult and Pediatric Patients. JAMA Neurol. 2020, 77, 257. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Li, Y.; Fu, Y.; Yang, L.; Su, L.; Shi, K.; Li, M.; Liu, Q.; Borazanci, A.; Liu, Y.; et al. Autoantibody to MOG Suggests Two Distinct Clinical Subtypes of NMOSD. Sci. China Life Sci. 2016, 59, 1270–1281. [Google Scholar] [CrossRef] [Green Version]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; for the PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. BMJ 2009, 339, b2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Li, Q.; Wang, T.; Fan, L.; Gao, L.; Huang, Z.; Lin, Y.; Xue, Q.; Liu, G.; Su, Y.; et al. Overlapping Syndrome of Anti-N-Methyl-D-Aspartate Receptor Encephalitis and Anti-Myelin Oligodendrocyte Glycoprotein Inflammatory Demyelinating Diseases: A Distinct Clinical Entity? Mult. Scler. Relat. Disord. 2021, 52, 103020. [Google Scholar] [CrossRef]
- Du, L.; Wang, H.; Zhou, H.; Chang, H.; Wei, Y.; Cong, H.; Xu, W.; Ma, Y.; Song, T.; Zhang, X.; et al. Anti-NMDA Receptor Encephalitis Concomitant with Myelin Oligodendrocyte Glycoprotein Antibody Diseases: A Retrospective Observational Study. Medicine 2020, 99, e21238. [Google Scholar] [CrossRef]
- Fan, S.; Xu, Y.; Ren, H.; Guan, H.; Feng, F.; Gao, X.; Ding, D.; Fang, F.; Shan, G.; Guan, T.; et al. Comparison of Myelin Oligodendrocyte Glycoprotein (MOG)-Antibody Disease and AQP4-IgG-Positive Neuromyelitis Optica Spectrum Disorder (NMOSD) When They Co-Exist with Anti-NMDA (N-Methyl-D-Aspartate) Receptor Encephalitis. Mult. Scler. Relat. Disord. 2018, 20, 144–152. [Google Scholar] [CrossRef]
- Hou, C.; Wu, W.; Tian, Y.; Zhang, Y.; Zhu, H.; Zeng, Y.; Peng, B.; Zheng, K.; Li, X.; Chen, W. Clinical Analysis of Anti-NMDAR Encephalitis Combined with MOG Antibody in Children. Mult. Scler. Relat. Disord. 2020, 42, 102018. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Kezuka, T.; Shikishima, K.; Yamagami, A.; Hiraoka, M.; Chuman, H.; Nakamura, M.; Hoshi, K.; Goseki, T.; Mashimo, K.; et al. Epidemiologic and Clinical Characteristics of Optic Neuritis in Japan. Ophthalmology 2019, 126, 1385–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Hernandez, E.; Guasp, M.; García-Serra, A.; Maudes, E.; Ariño, H.; Sepulveda, M.; Armangué, T.; Ramos, A.P.; Ben-Hur, T.; Iizuka, T.; et al. Clinical Significance of Anti-NMDAR Concurrent with Glial or Neuronal Surface Antibodies. Neurology 2020, 94, e2302–e2310. [Google Scholar] [CrossRef]
- Matsuda, R.; Kezuka, T.; Umazume, A.; Okunuki, Y.; Goto, H.; Tanaka, K. Clinical Profile of Anti-Myelin Oligodendrocyte Glycoprotein Antibody Seropositive Cases of Optic Neuritis. Neuro Ophthalmol. 2015, 39, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Kurimoto, T.; Murai, Y.; Ueda, K.; Sakamoto, M.; Chihara, N.; Yamada-Nakanishi, Y.; Nakamura, M. Efficacy for the Annual Relapse Rate after the Immunosuppressive Therapy in Patients Associated with Anti-AQP4 or Anti-MOG Antibody-Positive Optic Neuritis. J. Ophthalmol. 2020, 2020, 8871146. [Google Scholar] [CrossRef] [PubMed]
- Titulaer, M.J.; Höftberger, R.; Iizuka, T.; Leypoldt, F.; McCracken, L.; Cellucci, T.; Benson, L.A.; Shu, H.; Irioka, T.; Hirano, M.; et al. Overlapping Demyelinating Syndromes and Anti-N-Methyl-D-Aspartate Receptor Encephalitis: Anti-NMDAR Encephalitis. Ann. Neurol. 2014, 75, 411–428. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.B.; Zhou, L.; Zhang, Y.; Li, H.; Li, Y.; Huang, Y.; Wang, M.; Lu, C.; Lu, J.; et al. Encephalitis Is an Important Clinical Component of Myelin Oligodendrocyte Glycoprotein Antibody Associated Demyelination: A Single-center Cohort Study in Shanghai, China. Eur. J. Neurol. 2019, 26, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Woodhall, M.; Çoban, A.; Waters, P.; Ekizoğlu, E.; Kürtüncü, M.; Shugaiv, E.; Türkoğlu, R.; Akman-Demir, G.; Eraksoy, M.; Vincent, A.; et al. Glycine Receptor and Myelin Oligodendrocyte Glycoprotein Antibodies in Turkish Patients with Neuromyelitis Optica. J. Neurol. Sci. 2013, 335, 221–223. [Google Scholar] [CrossRef]
- Aoe, S.; Kokudo, Y.; Takata, T.; Kobara, H.; Yamamoto, M.; Touge, T.; Deguchi, K.; Masaki, T. Repeated Anti-N-Methyl-D-Aspartate Receptor Encephalitis Coexisting with Anti-Myelin Oligodendrocyte Glycoprotein Antibody-Associated Diseases: A Case Report. Mult. Scler. Relat. Disord. 2019, 35, 182–184. [Google Scholar] [CrossRef]
- Caparó-Zamalloa, C.; Álvarez-Toledo, K.; Yamunaque-Chunga, C.; Castro-Suarez, S.; Guevara-Silva, E.; Osorio-Marcatinco, V.; Meza-Vega, M. Autoimmune Neurology: Co-Occurrence of Anti-NMDAR Encephalitis and Anti-MOG Associated Disease, Report of a Case. J. Neuroimmunol. 2021, 358, 577663. [Google Scholar] [CrossRef]
- Cherian, A.; Divya, K.P.; Shetty, S.C.; Kannoth, S.; Thomas, B. Coexistent MOG, NMDAR, CASPR2 Antibody Positivity: Triumph over the Triumvirate. Mult. Scler. Relat. Disord. 2020, 46, 102468. [Google Scholar] [CrossRef] [PubMed]
- Cirkel, A.; Wandinger, K.-P.; Ditz, C.; Leppert, J.; Hanker, L.; Cirkel, C.; Neumann, A.; Brocke, J.; Höftberger, R.; Komorowski, L.; et al. Paraneoplastic Encephalomyeloradiculits with Multiple Autoantibodies against ITPR-1, GFAP and MOG: Case Report and Literature Review. Neurol. Res. Pract. 2021, 3, 48. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, J.; Takahashi, T.; Kaneko, K.; Atobe, Y.; Nakashima, I. Anti-NMDAR Encephalitis May Develop Concurrently with Anti-MOG Antibody-Associated Bilateral Medial Frontal Cerebral Cortical Encephalitis and Relapse with Elevated CSF IL-6 and CXCL13. Mult. Scler. Relat. Disord. 2021, 47, 102611. [Google Scholar] [CrossRef]
- Ji, S.; Liu, C.; Bi, Z.; Gao, H.; Sun, J.; Bu, B. Overlapping Syndrome Mimicking Infectious Meningoencephalitis in a Patient with MOG and GFAP IgG. BMC Neurol. 2021, 21, 348. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Sato, D.K.; Misu, T.; Kurosawa, K.; Nakashima, I.; Fujihara, K.; Aoki, M. Anti-N-Methyl-D-Aspartate Receptor Encephalitis with Multiphasic Demyelination. Ann. Neurol. 2014, 76, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Jiang, L. Viral Encephalitis Followed by Anti-NMDAR Encephalitis with Concomitant MOG Antibody-Positive Central Nervous System Demyelination in a Child. Neurol. Sci. 2020, 41, 2303–2305. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.C.; Marotta, D.A.; Kesserwani, H. Isolated Double-Positive Optic Neuritis: A Case of Aquaporin-4 and Myelin Oligodendrocyte Glycoprotein Antibody Seropositivity. Cureus 2021, 13, e15389. [Google Scholar] [CrossRef]
- Nan, D.; Zhang, Y.; Han, J.; Jin, T. Clinical Features and Management of Coexisting Anti-N-Methyl-D-Aspartate Receptor Encephalitis and Myelin Oligodendrocyte Glycoprotein Antibody–Associated Encephalomyelitis: A Case Report and Review of the Literature. Neurol. Sci. 2021, 42, 847–855. [Google Scholar] [CrossRef]
- Pérez, C.A.; Agyei, P.; Gogia, B.; Harrison, R.; Samudralwar, R. Overlapping Autoimmune Syndrome: A Case of Concomitant Anti-NMDAR Encephalitis and Myelin Oligodendrocyte Glycoprotein (MOG) Antibody Disease. J. Neuroimmunol. 2020, 339, 577124. [Google Scholar] [CrossRef]
- Ren, B.-Y.; Guo, Y.; Han, J.; Wang, Q.; Li, Z.-W. Case Report: Anti-NMDAR Encephalitis with Anti-MOG CNS Demyelination after Recurrent CNS Demyelination. Front. Neurol. 2021, 12, 639265. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, X.; He, Q.; Wang, R.; Lu, W. Co-Occurrence of Anti-N-Methyl-D-Aspartate Receptor Encephalitis and Anti-Myelin Oligodendrocyte Glycoprotein Inflammatory Demyelinating Diseases: A Clinical Phenomenon to Be Taken Seriously. Front. Neurol. 2019, 10, 1271. [Google Scholar] [CrossRef] [PubMed]
- Rojc, B.; Podnar, B.; Graus, F. A Case of Recurrent MOG Antibody Positive Bilateral Optic Neuritis and Anti-NMDAR Encephalitis: Different Biological Evolution of the Two Associated Antibodies. J. Neuroimmunol. 2019, 328, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Sarigecili, E.; Cobanogullari, M.D.; Komur, M.; Okuyaz, C. A Rare Concurrence: Antibodies against Myelin Oligodendrocyte Glycoprotein and N-Methyl-D-Aspartate Receptor in a Child. Mult. Scler. Relat. Disord. 2019, 28, 101–103. [Google Scholar] [CrossRef]
- Taraschenko, O.; Zabad, R. Overlapping Demyelinating Syndrome and Anti-N-Methyl-D-Aspartate Receptor Encephalitis with Seizures. Epilepsy Behav. Rep. 2019, 12, 100338. [Google Scholar] [CrossRef]
- Weiss, D.; Kertzscher, L.; Degering, M.; Wozniak, D.; Kluge, M. Anti-NMDA Receptor Encephalitis and Overlapping Demyelinating Disorder in a 20-Year Old Female with Borderline Personality Disorder: Proposal of a Diagnostic and Therapeutic Algorithm for Autoimmune Encephalitis in Psychiatric Patients “Case Report”. BMC Psychiatry 2021, 21, 355. [Google Scholar] [CrossRef]
- Zhou, J.; Tan, W.; Tan, S.E.; Hu, J.; Chen, Z.; Wang, K. An Unusual Case of Anti-MOG CNS Demyelination with Concomitant Mild Anti-NMDAR Encephalitis. J. Neuroimmunol. 2018, 320, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, J.B.; Li, H.; Li, X.; Huang, Y.; Wang, M.; Zhao, C.; Lu, J.; Lu, C.; Li, Y.; et al. Cerebral Cortical Encephalitis Followed by Recurrent CNS Demyelination in a Patient with Concomitant Anti-MOG and Anti-NMDA Receptor Antibodies. Mult. Scler. Relat. Disord. 2017, 18, 90–92. [Google Scholar] [CrossRef]
- Jurynczyk, M.; Geraldes, R.; Probert, F.; Woodhall, M.R.; Waters, P.; Tackley, G.; DeLuca, G.; Chandratre, S.; Leite, M.I.; Vincent, A.; et al. Distinct Brain Imaging Characteristics of Autoantibody-Mediated CNS Conditions and Multiple Sclerosis. Brain 2017, 140, 617–627. [Google Scholar] [CrossRef]
- Cornaby, C.; Gibbons, L.; Mayhew, V.; Sloan, C.S.; Welling, A.; Poole, B.D. B Cell Epitope Spreading: Mechanisms and Contribution to Autoimmune Diseases. Immunol. Lett. 2015, 163, 56–68. [Google Scholar] [CrossRef]
- Vanderlugt, C.L.; Miller, S.D. Epitope Spreading in Immune-Mediated Diseases: Implications for Immunotherapy. Nat. Rev. Immunol. 2002, 2, 85–95. [Google Scholar] [CrossRef]
- Dalmau, J.; Lancaster, E.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R. Clinical Experience and Laboratory Investigations in Patients with Anti-NMDAR Encephalitis. Lancet Neurol. 2011, 10, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Dalmau, J.; Tüzün, E.; Wu, H.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic Anti-N-Methyl-D-Aspartate Receptor Encephalitis Associated with Ovarian Teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | |
|---|---|
| Age at onset, median (range), y | 21 (2~63) |
| Female, n (%) a | 29 (40.28) |
| Titer of serum MOG-IgG b | 1:100 (1:10~1:16,384) |
| Titer of CSF NMDAR-IgG c | 1:32 (1:1~1:320) |
| Follow-up duration, median (range), mod | 15 (2~144) |
| mRS score ≤ 2 at last follow-up, n (%) e | 26 (92.86) |
| Features | |
|---|---|
| Age at onset, median (range), y a | 35 (15–66) |
| Female, n (%) b | 23 (95.8) |
| Disease duration, median (range), y c | 4 (2–11) |
| EDSS score at last follow-up d | 8 (5–9) |
| Attack number e | 6 (1–10) |
| OCB, n (%) f | 1 (8.3) |
| No./Age/Gender | Coexisting Antibodies | Clinical Manifestation | Imaging Features |
|---|---|---|---|
| 1/48/F (13) | CASPR2-IgG | Decreased vision, dizziness, speech disorder, gait instability, urinary incontinence, psychiatric symptoms | Hyperintensities in cortex, cerebral peduncle, brainstem, thalamus, corpus callosum, cervical and thoracic spinal cord |
| 2/10/M (11) | CASPR2-IgG | Ascending paralysis, intractable seizures | ADEM-like lesions involving bilateral hemisphere, brainstem, and cerebral peduncle |
| 3/30/M (37) | NMDAR-IgG and CASPR2-IgG | Headache, psychological and behavioral abnormalities, memory loss, cerebellar dysarthria, spastic ataxia | Hyperintensities in bilateral cingulate gyri, hippocampus, pulvinar; patchy perivascular and subpial enhancement over pons, cerebellar peduncle, cerebellar folia, midbrain, and cingulate gyri |
| 4/59/M (11) | GABAA-R-IgG | Focal seizures, encephalopathy | Hyperintensities in bilateral temporal lobe |
| 5/55/F (11) | LGI1-IgG | NA | NA |
| 6/20/M (12) | GFAP-IgG | Decreased vision, diplopia, nystagmus, dizziness, hemiplegia, Romberg’s sign | Swelling of bilateral ON; hyperintense patchy lesions in cerebellum, brachium pontis, and temporal lobe |
| 7/23/F (42) | GFAP-IgG | Fever, headache, vomiting, convulsion, Kernig sign | Diffuse leptomeningeal enhancement; asymmetric hyperintense signal in cerebellum and corona radiata, radial enhancement patterns extending outward from the ventricles |
| 8/27/F (11) | AQP4-IgG and NMDAR-IgG | Optic neuritis, cervical LETM | Lesions in temporal lobe, thalamus, optic tract and chiasm, spinal cord |
| 9/33/M (11) | GFAP-IgG and NMDAR-IgG | First attack: multifocal meningoencephalitis; second attack: cervical LETM | First attack: T2 hyperintensity and leptomeningeal enhancement along left temporal, frontal, and parietal cortexes; second attack: hyperintensities in basal ganglia, cerebellar peduncle, and spinal cord |
| 10/44/F (40) | GFAP-IgG and ITPR-1-IgG | Fever, nausea, vomiting, paraplegia, ataxia, nystagmus, urinary retention, respiratory paralysis | Signs of meningitis, cortical and subcortical lesions within parietooccipital cortex with diffuse restriction, edema of medulla oblongata |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, C.; Liu, P.; Zhao, D.; Ding, J.; Zhang, G.; Li, H.; Guo, J. Coexistence of Myelin Oligodendrocyte Glycoprotein Immunoglobulin G and Neuronal or Glial Antibodies in the Central Nervous System: A Systematic Review. Brain Sci. 2022, 12, 995. https://doi.org/10.3390/brainsci12080995
Zhao C, Liu P, Zhao D, Ding J, Zhang G, Li H, Guo J. Coexistence of Myelin Oligodendrocyte Glycoprotein Immunoglobulin G and Neuronal or Glial Antibodies in the Central Nervous System: A Systematic Review. Brain Sciences. 2022; 12(8):995. https://doi.org/10.3390/brainsci12080995
Chicago/Turabian StyleZhao, Cong, Pei Liu, Daidi Zhao, Jiaqi Ding, Guangyun Zhang, Hongzeng Li, and Jun Guo. 2022. "Coexistence of Myelin Oligodendrocyte Glycoprotein Immunoglobulin G and Neuronal or Glial Antibodies in the Central Nervous System: A Systematic Review" Brain Sciences 12, no. 8: 995. https://doi.org/10.3390/brainsci12080995