
Abstract
Background
To report the clinical and laboratory characteristics, clinical courses, and outcomes of Mayo Clinic, Rochester, MN, ICU-managed autoimmune encephalitis patients (January 1st 2003–December 31st 2012).
Methods
Based on medical record review, twenty-five patients were assigned to Group 1 (had ≥1 of classic autoimmune encephalitis-specific IgGs, n = 13) or Group 2 (had ≥3 other characteristics supporting autoimmunity, n = 12).
Results
Median admission age was 47 years (range 22–88); 17 were women. Initial symptoms included ≥1 of subacute confusion or cognitive decline, 13; seizures, 12; craniocervical pain, 5; and personality change, 4. Thirteen Group 1 patients were seropositive for ≥1 of VGKC-complex-IgG (6; including Lgi1-IgG in 2), NMDA-R-IgG (4), AMPA-R-IgG (1), ANNA-1 (1), Ma1/Ma2 antibody (1), and PCA-1 (1). Twelve Group 2 patients had ≥3 other findings supportive of an autoimmune diagnosis (median 4; range 3–5): ≥1 other antibody type detected, 9; an inflammatory CSF, 8; ≥1 coexisting autoimmune disease, 7; an immunotherapy response, 7; limbic encephalitic MRI changes, 5; a paraneoplastic cause, 4; and diagnostic neuropathological findings, 2. Among 11 patients ICU-managed for ≥4 days, neurological improvements were attributable to corticosteroids (5/7 treated), plasmapheresis (3/7), or rituximab (1/3). At last follow-up, 10 patients had died. Of the remaining 15 patients, 6 (24 %) had mild or no disability, 3 (12 %) had moderate cognitive problems, and 6 (24 %) had dementia (1 was bed bound). Median modified Rankin score at last follow-up was 3 (range 0–6).
Conclusions
Good outcomes may occur in ICU-managed autoimmune encephalitis patients. Clinical and testing characteristics are diverse. Comprehensive diagnostics should be pursued to facilitate timely treatment.
Similar content being viewed by others


Introduction
In office-based practice, patients with autoimmune encephalopathies usually present with subacute onset and rapid progression of mental decline and intermittent partial seizures. The differential diagnosis is usually limited to metabolic and rapidly progressive neurodegenerative disorders [1, 2]. In many instances, improvements often coincide with immunotherapy, and good or excellent outcomes seem to be the norm [3–6]. Autoimmune encephalitis occurring in critically ill patients who present to hospital is characterized by grossly altered sensorium, the need for airway protection and intubation, and intractable seizures [7, 8]. The need for intensive care unit (ICU) admission may predict poor outcome [6].
Certain autoimmune encephalitides are recognizable by their classic serological findings. Examples include Lgi1 antibody-associated limbic encephalitis and NMDA receptor encephalitis [9–12]. In our experience of diagnosing and treating consecutively referred critically ill autoimmune encephalitis patients, clinical and testing findings are diverse, and do not always fall under the rubric of these classic disorders. In the ICU setting, clinical courses have multiple neurological and medical complications. Neurological disorders are often refractory to immunotherapies, albeit the final outcome may still be favorable.
Herein, we describe the clinical and other diagnostic features that facilitated recognition of autoimmune encephalitis on our ICU services at Mayo Clinic Rochester (2003–2012). In addition, we describe responses to treatment, complications, and outcomes.
Methods
The Mayo Clinic institutional review board approved this study, (12-09670).
Participants
The methods are described in detail in the Supplementary Appendix and outlined in Fig. 1. We searched for all adult encephalitis patients admitted to any ICU in the Mayo Clinic Rochester hospitals from January 1st 2003 to December 31st 2012 [13]. Among 635 patients with encephalitis or encephalopathy that had MRI head imaging, a CSF evaluation and at least one serum or CSF specimen sent to the Mayo Clinic Neuroimmunology Laboratory, 68 were initially included as having autoimmune encephalopathies or uncertain diagnoses. Their archived specimens were re-evaluated to ensure that comprehensive antibody testing was undertaken, as previously described [14, 15]. Forty-three of 68 patients were excluded (Supplementary Appendix). The remaining 25 patients were either

Algorithm of process of patient inclusion, Groups 1 and 2
-
1)
Group 1 Thirteen patients with ≥1 neural-specific IgGs classically associated with autoimmune encephalitis detected in serum, CSF or both.
-
2)
Group 2 Twelve patients who did not fulfill Group 1 criteria but had at least 3 other findings supportive of an autoimmune diagnosis: classic accompanying clinical or radiological autoimmune phenotype, neuropathology consistent with an autoimmune or paraneoplastic cause, ≥1 antibody not meeting criteria for Group 1; an inflammatory spinal fluid; ≥1 coexisting autoimmune disease; a contemporaneous cancer diagnosis indicative of a paraneoplastic cause and documented neurological improvement after a course of immunotherapy. An inflammatory CSF was determined by the presence of at least 2 of protein ≥70 mg/dL, >5 white cells, ≥4 CSF-exclusive oligoclonal bands, an elevated IgG synthesis rate, and an elevated IgG index [4].
Statistical Comparison
Age, male sex, symptom duration before treatment, prior history of autoimmune encephalitis, seizures on EEG, status epilepticus, a paraneoplastic cause, an inflammatory CSF, detection of a neuronal nuclear or cytoplasmic paraneoplastic antibody (such as ANNA-1 or Ma2), and sepsis were assessed as predictors of (1) physician-reported improvement with immunotherapy and (2) poor outcome (defined as final modified Rankin score of >3) using Fisher exact test or Chi square test for dichotomous variables and Wilcoxon test for continuous variables.
Results
Demographic, clinical, and paraclinical data for the 25 patients are described in Tables 1 and 2.
Demographic Information
Median age at ICU admission was 47 years (range 22–88); 17 were women. Seventeen patients were admitted to the Neurosciences ICU (NICU), six were evaluated by a neurointensivist consult service in the medical ICU, and 2 had both NICU and medical ICU admissions during hospitalization. Twelve patients had direct admission to the ICU from the Emergency Room, and the remaining 13 were first admitted to the general neurology service. Ten patients (40 %) had a diagnosis of autoimmune encephalitis prior to admission to the NICU. Alternative diagnoses (where stated) in 7 were viral encephalitis, 4; rapidly progressive neurodegenerative dementia, vasculitis, and vestibular neuritis, 1 each. All 25 patients had been diagnosed with autoimmune encephalitis by the last point of follow-up. Median duration of symptoms prior to ICU admission was 4.5 weeks (range 1–90). Median duration of stay in ICU was 3 days (range 1–135).
Clinical Presentations
Initial symptoms included one or more of confusion or subacute cognitive decline, 13; seizures, 12; head or neck pain, 5; personality or behavioral change, 4; myoclonic jerking, 2; ataxic symptoms, 2; coma, sensory symptoms, aphasia, dysphagia and dysarthria, and fever, 1 patient for each. Indications for ICU admission were one or more of seizures or status epilepticus, 13; respiratory failure, 5 (including 3 with pulmonary embolism); agitation, 3; coma, 3; systemic infection, 2; hemorrhage, 2 (retroperitoneal, 1; upper gastrointestinal, 1); labile blood pressure or heart rate, 2; and management of raised intracranial pressure secondary to cerebral edema, 1. Eight cancers detected contemporaneous to the neurologic disorder in 7 patients (28 %) were adenocarcinomas, 4 (lung, 2; breast, 1; esophageal, 1); small-cell carcinoma of lung, 1; non-small-cell carcinoma of lung, 1; metastatic spindle cell carcinoma, 1; and myeloma, 1.
Coexisting Autoimmune Diseases
One or more coexisting autoimmune diseases were present in 12 patients (48 %). These were thyroid disease, 6 (Hashimoto thyroiditis, 5; Graves’ disease, 1); Sjogren syndrome, 2; ankylosing spondylitis, 2; rheumatoid arthritis, 1; vitiligo, 1; systemic lupus erythematosus, 1; and pernicious anemia, 1.
Test Results
Brain Imaging
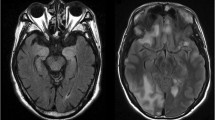
MRI head imaging (Fig. 2) showed T2-signal abnormalities indicative of inflammation in 14 (56 %; limbic, 7; extra-limbic, 6; both limbic and extra-limbic, 1); non-specific leukoaraiosis in 4, normal findings in 4; and atrophy in 3. One patient with coma had both findings consistent with limbic encephalitis and small cerebellar brain metastases. One patient had enhancement post-gadolinium administration and three had hyperintensities on diffusion-weighted imaging, including changes compatible with posterior reversible encephalopathy syndrome (PRES) in 1 patient. One patient had areas of focal hemorrhage and necrosis in bilateral frontoparietal regions. The 4 NMDA-R-IgG positive patients had normal or non-specifically abnormal-appearing MRI scans.

Selected MRI head images from autoimmune encephalitis patients managed in the Neurosciences ICU. MRI sequences were FLAIR (axial [a, b, f, and h] or coronal [c and g]), susceptibility-weighted (axial, d) and T1, post-gadolinium (coronal, e). Patient 2 a with NMDA-R encephalitis had a normal head MRI but fatal disease. Patient 3 b with Ma1 and Ma2 antibodies had classic limbic encephalitic-appearing hyperintense mesial temporal lobes bilaterally. Patient 15 (c–e) who was encephalitis antibody negative, but had 3 systemic autoimmune diseases had diffuse frontoparietal white matter abnormalities (c), hypointensities consistent with hemorrhage (d), and enhancement (e). Patient 16 with Sjogren syndrome had parietal and occipital T2 hyperintensities consistent with PRES (f). Patient 24 with ankylosing spondylitis and autoimmune diabetes mellitus had abnormal hippocampal T2 signal (g) that extended posteriorly (h)
Electroencephalography
Twenty-four patients had electroencephalogram (EEG) performed; all but one were abnormal. Twenty-two had generalized slowing. Seven patients had electrographic seizures (28 %) that were focal (4) or multifocal (3). None had primary generalized seizures. Three had additional focal slowing without epileptiform discharges. Of 10 patients with focal EEG abnormalities, 5 had temporal localization, four had frontotemporal localization, and one had both left temporoparietal and right parasagittal seizure discharges.
CSF
Of 25 patients, 20 had findings indicative of CNS inflammation (80 %). Findings included ≥5 white cells, 12; protein ≥70 mg/dL, 9; ≥4 oligoclonal bands, 8; elevated IgG synthesis rate, 4; and elevated IgG index, 4.
Classification
Based on serological findings, patients were divided into 2 groups. Group 1 patients had one or more neural-specific IgGs classically associated with autoimmune encephalitis detected in serum or CSF. Group 2 patients had other findings supportive of an autoimmune diagnosis.
Group 1
The neural-specific IgGs detected in serum alone, CSF alone, or both of 13 patients included VGKC-complex IgGs, 6 (including Lgi1-IgG in 2); NMDA-R-IgG, 4; AMPA-R-IgG, 1; ANNA-1, 1; Ma1 and Ma2 antibody, 1; and PCA-1, 1. Patient 10 had coexisting NMDA-R-IgG and VGKC-complex IgG (Table 1 and eTable 1). She was subsequently found to have small-cell carcinoma of lung.
The serological diagnosis was made at the time of hospitalization in 11 of 13 patients. During re-evaluation of historic serum and CSF specimens for this study, encephalitis-specific IgGs, described in the literature after the patients’ clinical evaluations, were detected in 2 patients: NMDA-R-IgG (Patient 2) and AMPA-R-IgG (Patient 13). Patient 2, a 22-year-old woman, was neural autoantibody seronegative at the time of evaluation in 2005. She presented with agitation and catatonic behavior, followed by coma, and was admitted to the NICU. She received electroconvulsive therapy without improvement. A diagnosis of likely autoimmune encephalitis was made and plasma exchange alone was initiated. She did not improve. After 79 days in the NICU, supportive care was withdrawn, and she died a short time later. Patient 13, a 48-year-old woman, had Sjogren syndrome and hypothyroidism, developed encephalopathy, but was not recognized as having an autoimmune disorder for 1 year prior to transfer to our hospital in 2010. On admission, she was found to have adenocarcinoma of breast and was diagnosed with paraneoplastic limbic encephalitis. She had modest neurological improvements attributable to plasma exchange, but not corticosteroids or IVIg.
Group 2
Twelve patients (48 %) did not have a neural-specific IgG classically associated with autoimmune encephalitis detected in serum or CSF (Table 2 and eTable 2). These patients had 3 or more other findings supportive of an autoimmune diagnosis (median 4; range 3–5): ≥1 coexisting neural or non-neural antibody detected in serum or CSF not meeting criteria for Group 1, 9; an inflammatory CSF, 8; ≥1 coexisting autoimmune disease, 7; an immunotherapy response, 7; classic limbic encephalitic changes on MRI, 5; a paraneoplastic cause (concomitant systemic cancer diagnosis), 4; classic neuropathology consistent with an autoimmune or paraneoplastic cause, 2; and additional classic accompanying autoimmune neurological phenotype (opsoclonus-myoclonus syndrome), 1.
Five patients had neural antibodies detected: low positive values of GAD65-IgG, 2; unclassified neural antibodies, 2; and muscle AChR antibody, 1 (did not have myasthenia gravis). One or more non-neural antibodies detected in 6 patients were against: nuclear antigens, 3 (extractable nuclear antigens [ENA], 2; not differentiated, 1); thyroid peroxidase, 3; neutrophilic antigens, (proteinase 3, 1; myeloperoxidase, 1); and cyclic citrullinated peptides, 1. ENA positivities included ribonuclear protein, 1 and sjogren syndrome antigen (SS-A), 1. One had rheumatoid factor.
Treatment and Complications
Fourteen patients (56 %) had a short ICU stay (1–3 days) before returning to the general neurology service. The remaining 11 patients (44 %) had a prolonged ICU stay, median 14 days (range 4–90; Table 3). Neurological or medical complications arose in all patients having a prolonged ICU stay (Table 4).
Immunotherapies and Oncological Therapies
Nine of 11 patients with ICU stay longer than 3 days received immunotherapy during that stay (Table 3). These treatments included corticosteroids, 7 (5 improved); plasma exchange, 7 (3 improved); and IVIg, 3 (none improved). One of 3 patients who received rituximab improved. After dismissal from the hospital, 3 patients who responded to corticosteroids achieved successful remission maintenance with a steroid-sparing immunosuppressant (azathioprine, 2; mycophenolate mofetil, 1). Three patients with paraneoplastic neurological disorders received oncological therapy (surgery, 2; chemotherapy, 1); none had neurological improvements. Patient 14 had metastatic non-small-cell lung carcinoma, pneumonia, and acute renal failure, in addition to limbic encephalitis and received palliative care only.
Complications in the ICU and Their Management
Neurological complications that arose in the ICU included paroxysmal sympathetic hyperactivity (hyperhidrosis and tachycardia, 5), refractory seizures (5), severe agitation (3), and hyperkinetic movement disorders (5; dyskinesias, 2; myoclonus, 2; chorea, 1). Gabapentin 900–3600 mg per day in three divided doses suppressed sympathetic hyperactivity in the 3 patients treated. All 5 patients with status epilepticus in the ICU required 3 or more antiepileptic drugs to control seizures, including levetiracetam (5), fosphenytoin (5), midazolam (4), lacosamide (2), valproic acid (2), pentobarbital (2), phenobarbital (1), and ketamine (1). Agitation was initially controlled with combinations of midazolam (3), dexmedetomidine (3), and propofol (1), and then treatment was transitioned from IV to one or more oral agents (quetiapine [2]; clonazepam [2]). Dyskinetic movements in 3 patients with NMDA-R encephalitis were only suppressible with propofol (2) or pentobarbital (1). Patient 21 required midazolam and vecuronium to suppress large-amplitude myoclonus.
Medical complications included anemia (8), sepsis (8), pulmonary edema (7), venous thrombosis (6), significant electrolyte or acid–base disturbances (6), cardiac dysrhythmia (4), acute renal failure (4), heparin-induced thrombocytopenia syndrome (3), hypotension (3), malignant hypertension (2), allergic drug reaction (2; phenytoin and opiates, 1; lacosamide, 1), anasarca (2), bronchorrhea (1), and adrenal insufficiency (1).
Outcomes
At last follow-up, 10 patients had died (40 %), 6 during hospitalization and 4 after discharge (median 5 months; range 1–55 months), and thus, the median duration from admission to ICU until last follow-up was short (4 months; range 0.25–60). Of the 6 who died during hospitalization, 5 were transitioned to palliative care and one had sudden cardiac death. Disorders contributing to death for patients transitioned to palliative care were severe encephalopathy, 3; status epilepticus, 2; sepsis, 2; renal failure, 2; and metastatic cancer, 1. Of 4 patients who died after hospitalization, 3 had paraneoplastic limbic encephalitis and active cancer diagnoses but were lost to follow-up at the time of death. The fourth (Patient 3) had a severe, intractable brainstem and limbic encephalitis (Ma1 and Ma2 antibody seropositive), without cancer detected, and died suddenly in his sleep. Of the remaining 15 patients, 6 (24 %) had mild or no disability, 3 (12 %) had moderate cognitive problems, and 6 (24 %) had dementia (1 was bed bound). Median modified Rankin score at last follow-up was 3 (range 0–6).
Outcomes for the 11 patients with prolonged ICU stay (4 or more days) are described in Table 4; in this subgroup, 5 of these patients died during the acute hospitalization, 2 died after hospitalization, and all 4 survivors made a good recovery (all followed for at least 18 months). No statistically significant predictors of immunotherapy response or poor outcome were identified. There was a trend to statistical significance for shorter symptom duration prior to treatment being predictive of immunotherapy response (mean duration of symptoms in days for responders, 65; SD ± 85; mean duration of symptoms in days for non-responders, 377; SD ± 761), p = 0.05 (Wilcoxon Rank-Sum test).
By comparison, 28 patients from our Autoimmune Neurology Clinic experience (2009–2014) who did not require ICU admission, but otherwise met criteria for Group 1 (13) or Group 2 (15) inclusion, almost always improved with immunotherapy (21 patients) and had an mRS of ≤3 (25), and survived in all cases.
Discussion
Due to the nature of its clinical course, patients with autoimmune encephalitis managed in our ICUs often had a poor prognosis despite the timely use of immunotherapy and prolonged critical care. Yet, good recovery was achieved by a sizeable minority of our patients. In just over half, patients were diagnosable by detection of a well-characterized neural autoantibody in serum or CSF. For the remainder, a search for other clues was required to yield diagnoses. The frequency of encephalitis antibody seronegativity in our cohort was similar to a recent cohort of children with autoimmune encephalitis diagnosed in the United Kingdom (44 %) [16]. These experiences underline the importance of a comprehensive diagnostic approach; a detailed clinical history and diagnostic evaluation, including antibody testing of serum and CSF [17].
For some patients (Group 1), the diagnosis became obvious because of the encephalitis-specific antibody detected, but in many other patients, additional data, both clinical (systemic autoimmune and cancer diagnoses) and paraclinical (radiological, serological, and CSF findings), had to be pooled to make the diagnosis (Group 2). MR head imaging did not show inflammatory-appearing abnormalities in 40 %, and just two of our patients exhibited contrast enhancement. EEG was almost always abnormal, but those abnormalities alone were not specific for neurological autoimmunity. Non-classic extra-limbic temporal or extra-temporal radiological and EEG changes sometimes occurred also [15]. CSF usually (but not always) proved diagnostically helpful, with inflammatory features detected in 80 %. In some Group 2 patients, the suspected diagnosis was uncertain until confirmed by an observed immunotherapy response. The NMDA-R encephalitis cases encountered predated the description of ‘extreme delta brush’ which may be a specific EEG finding for that disorder [18].
Early treatment may be a factor influencing better outcome [6]. Some patients improve greatly with initial intravenous methylprednisolone, but others require continued high doses of corticosteroid as well as other treatment modalities (plasma exchange or IVIg). Others still may be candidates for immunosuppression but are also at high risk for sepsis, which may portend a worse prognosis. Patients 2 and 13 were only determined to have encephalitis-specific IgGs retrospectively in the course of this study. Despite additional clues at the illness onset, the recognition of their disorders as autoimmune and treatment came late because of the lack of these classic biomarkers.
In addition to diagnostic importance, antigen specificity of neural autoantibodies may have prognostic significance. Some autoantibodies target neuronal intracellular nuclear or cytoplasmic antigens in antibody assays, but are unlikely to be pathogenic [19]. Accompanying disorders are likely cytotoxic T lymphocyte-mediated, and are usually not immunotherapy responsive. For example, Patient 6 with ANNA-1 and small-cell carcinoma progressed rapidly despite steroid and IVIg treatment. Other autoantibodies targeting plasma membrane protein antigens, are likely pathogenic and accompanying neurological disorders often (but not always) improve with immunotherapy [20]. Five of 6 patients seropositive for VGKC-complex IgGs improved with immunotherapy.
Outcomes among NMDA-R encephalitis patients varied. Patient 10 with NMDA-R-IgG coexisting with VGKC- complex IgG had a prolonged ICU stay (33 days) but achieved considerable and sustained improvement with corticosteroids and rituximab. In contrast, Patient 12, with the same diagnosis, had ongoing neurological progression and died despite maximum treatment with immunosuppression. This experience is distinct from a previous study which reported a favorable outcome in all 7 NMDA-R-IgG patients admitted to ICU [21]. Consistent with our own experience, a small case series (n = 6) of patients with NMDA receptor encephalitis reported good outcome in 3 and severe deficits or death in the other 3 patients [22] Long-lived plasma cells in the CNS might elude the effects of immunotherapy in some NMDA-R encephalitis patients [23].
Our series represents the extreme of severity in the autoimmune encephalitis spectrum. Refractory seizures, status epilepticus, and systemic complications were frequent. Conversely, intracranial hypertension was rare. The most common neurological complications, including refractory epilepsy, paroxysmal sympathetic hyperactivity, and severe agitation with hyperkinesis, were difficult to control, but often treatable. Strict monitoring for systemic complications (sepsis, heart rate, and blood pressure fluctuations, ileus caused by dysautonomia, anemia, and immobility-related venous thromboembolism) is essential to optimize outcomes.
Patients in this series often died or were left with mostly cognitive disability. Still, nearly a third experienced excellent recovery. In a previous study, critically ill patients with autoimmune disorders had better prognosis than other forms of encephalitis [24]. The percentage of patients with autoimmune encephalitis who achieved good outcome was not provided and only 6 of 16 patients classified as autoimmune had an antibody detected, making comparison with our cohort difficult. Therefore, although outcomes may be unfavorable, aggressive treatment and prolonged supportive care should be pursued because good recovery is possible.
Our study has the limitations inherent to retrospective studies. There are no FDA-approved treatments for autoimmune encephalitis and standardized treatment protocols had not been established during the epoch of this particular study. While no patient received cyclophosphamide or rituximab as first line therapy, steroids, IVIg, and plasma exchange were used in a variety of orders and combinations. Responses to therapy were based on physician reports documented in the medical records. The range of improvements varied, but in each patient was largely commensurate with the final outcome. The small cohort size limited our analysis of outcome predictors. Furthermore, it is possible that some radiologic and CSF abnormalities were caused by status epilepticus alone. When interpreting outcomes, it should be recalled that some patients were transitioned to palliative care because of overall neurological or oncological prognosis. Similarly, in cases with better prognosis, the follow-up may have been insufficient to evaluate full therapy response.
It is important to consider an autoimmune cause when evaluating any patient presenting to the ICU with subacute severe encephalopathy or encephalitis, even if not syndromically classic. Where appropriate, evaluations for occult cancer performed. We recommend treating with one or more of steroids, IVIg, or plasma exchange, before neuronal antibody test results (which are frequently negative) are available. In ICU patients with established autoimmune encephalopathy diagnoses with suboptimal improvements from early treatment, immunosuppression with rituximab, or cyclophosphamide treatment may bring about further neurological improvements but the risk of infection is high.
References
Kelley BJ, Boeve BF, Josephs KA. Young-onset dementia: demographic and etiologic characteristics of 235 patients. Arch Neurol. 2008;65:1502–8.
Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol. 2011;70:437–44.
McKeon A. Immunotherapeutics for autoimmune encephalopathies and dementias. Curr Treat Options Neurol. 2013;15:723–37.
Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc. 2010;85:881–97.
Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127:701–12.
Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12:157–65.
Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005;58:594–604.
McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2013;3:53–64.
Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9:776–85.
Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69:892–900.
Klein CJ, Lennon VA, Aston PA, et al. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol. 2013;70:229–34.
Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13:167–77.
Herasevich V, Pickering BW, Dong Y, Peters SG, Gajic O. Informatics infrastructure for syndrome surveillance, decision support, reporting, and modeling of critical illness. Mayo Clin Proc. 2010;85:247–54.
O’Toole O, Lennon VA, Ahlskog JE, et al. Autoimmune chorea in adults. Neurology. 2013;80:1133–44.
Quek AM, Britton JW, McKeon A, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol. 2012;69:582–93.
Hacohen Y, Wright S, Waters P, et al. Paediatric autoimmune encephalopathies: clinical features, laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens. J Neurol Neurosurg Psychiatry. 2012;. doi:10.1136/jnnp-2012-303807.
McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76:1108–10.
Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D. Extreme delta brush: a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology. 2012;79:1094–100.
McKeon A, Pittock SJ. Paraneoplastic encephalomyelopathies: pathology and mechanisms. Acta Neuropathol. 2011;122:381–400.
McKeon A, Martinez-Hernandez E, Lancaster E, et al. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol. 2013;70:44–50.
Pruss H, Dalmau J, Harms L, et al. Retrospective analysis of NMDA receptor antibodies in encephalitis of unknown origin. Neurology. 2010;75:1735–9.
Davies G, Irani SR, Coltart C, et al. Anti-N-methyl-D-aspartate receptor antibodies: a potentially treatable cause of encephalitis in the intensive care unit. Crit Care Med. 2010;38:679–82.
Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology. 2011;77:589–93.
Thakur KT, Motta M, Asemota AO, et al. Predictors of outcome in acute encephalitis. Neurology. 2013;81:793–800.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Alejandro A. Rabinstein receives royalties from Elsevier and Oxford University Press for authored books and research support from DJO Global. Sara E. Hocker serves on the Data Safety Monitoring Board for Sage Therapeutics. Sean J. Pittock has received no royalties to date but may accrue revenue for patents relating to AQP4 antibodies for diagnosis of neuromyelitis optica and AQP4 autoantibody as a cancer marker. He receives research support from the Guthy-Jackson Charitable Foundation, Alexion Pharmaceuticals, Inc. and the National Institutes of Health (RO1 NS065829). Eelco F.M Wijdicks serves as the editor-in-chief of Neurocritical Care and receives royalties from books published by Oxford University Press, Springer and CRC press. Andrew McKeon receives research support from MedImmune, Inc. Manoj K. Mittal reports no disclosures.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mittal, M.K., Rabinstein, A.A., Hocker, S.E. et al. Autoimmune Encephalitis in the ICU: Analysis of Phenotypes, Serologic Findings, and Outcomes. Neurocrit Care 24, 240–250 (2016). https://doi.org/10.1007/s12028-015-0196-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-015-0196-8