Introduction
Neuromyelitis optica (NMO) and NMO spectrum disorders (NMOSD) are clinical syndromes traditionally defined by the presence of myelitis and optic neuritis (ON). Aquaporin-4 (AQP4) is a water channel that is heavily expressed on astrocyte foot processes in the optic nerves, brainstem, and spinal cord. Antibodies to AQP4 are directly pathogenic and serve as a highly specific diagnostic biomarker for NMO, which is technically an autoimmune astrocytopathy.1 They are detectable in 60% to 80% of patients with NMOSD. Lesions that are positive for AQP4 antibodies (AQP4+), which have a prominent perivascular complement deposition, are pathologically distinct from lesions of multiple sclerosis (MS). Among patients with an NMOSD phenotype who are seronegative for antibodies to AQP4 (AQP4-), as many as 42% have detectable serum antibodies to myelin oligodendrocyte glycoprotein (MOG) (See Box).3 The prevalence of NMOSD is roughly 4 to 10 per 100,000, with higher rates among those of African or Asian descent; onset occurs throughout the lifespan and 5 to 10 times more often in women than men.4,5
Clinical Manifestations
Most patients with NMOSD have a relapsing course with attacks that have a temporal profile similar to a demyelinating episode of MS. Relapses in patients with AQP4+ NMOSD are more likely to be severe and disabling compared with MS relapses. The cardinal clinical features of NMOSD include episodes of optic neuritis, acute myelitis, and brainstem syndromes. Other core clinical features that occur less often include symptomatic narcolepsy, acute diencephalic syndrome, or acute cerebral syndromes.6
Optic Neuritis
Inflammatory ON typically presents as acute to subacute monocular vision loss with impaired visual acuity that may be accompanied by color desaturation, a relative afferent pupillary defect, and/or optic disc edema. Clinical features that suggest NMOSD (or MOG antibody-associated disorder) include simultaneously bilateral ON and severe vision loss (acuity 20/200 or worse).7,8
Brainstem Syndromes
The area postrema (AP) is an emetic reflex center located in the floor of the fourth ventricle that modulates a number of physiologic processes such as hiccups and emesis. The AP has been proposed as the initial site of entry for pathogenic AQP4 antibodies because AP capillaries lack tight junctions and AP astrocytes express an abundance of AQP4.9
Area postrema syndrome (APS) occurs commonly in AQP4+ NMOSD consequent to inflammatory lesions in the AP.10 In contrast to ON or myelitis, AP lesions lack demyelination or necrosis, which may in part explain the nearly universal complete remission of symptoms after an attack. Proposed APS diagnostic criteria include acute or subacute nausea, vomiting, or hiccups lasting for at least 48 hours without response to treatment or evidence of other etiologies (eg, hyponatremia, CNS structural lesions, and gastrointestinal or migrainous phenomena).11 Dorsal medulla lesions on MRI can facilitate early diagnosis. An attack of APS may serve as warning of an impending attack of ON or myelitis as most occurrences (58%-68%) precede inflammatory involvement elsewhere.11 Other brainstem syndromes sometimes encountered in patients with NMOSD include oculomotor abnormalities, hearing loss, nuclear facial nerve palsy, vestibular dysfunction, and other cranial nerve abnormalities.12
Myelitis
Considered a hallmark of NMOSD, longitudinally extensive transverse myelitis (LETM) is defined as myelitis spanning at least 3 spinal segments. Neurologists should be aware that short-segment myelitis is also common in NMOSD; neglecting to consider this is associated with delay in diagnosis and treatment.13 The anterolateral cord syndrome with prominent weakness and spinothalamic tract deficits is a typical but not exclusive pattern.
Pregnancy
Relapse rates in women with NMOSD do not change during pregnancy in contrast to women with MS, who have a reduced relapse rate during pregnancy;14 relapse risk increases during the first 3 months postpartum in both women with NMOSD and MS, however.15 The placenta expresses AQP4, which may in part explain why preeclampsia, intrauterine growth restriction, and miscarriage are more common in women with NMOSD. Antibodies to AQP4 are transferred across the placenta but do not appear to be pathogenic to the fetus.
Children
Although median age of onset is 30 to 40 years, 3% to 5% of patients with NMOSD present as children, which can be clinically challenging.16 The most common presenting features of NMOSD in children include visual, motor, and constitutional symptoms (ie, vomiting, fever, seizures). A single study reported a mean annualized relapse rate (ARR) of 0.6 in pediatric-onset NMOSD compared with 1.0 in adult-onset NMOSD; time to disability is longer in patients with pediatric-onset, largely explained by the severity of a first episode of myelitis in adults. Time to first treatment was significantly longer in children compared with adults (13.1 vs 3.4 years). Children with acute disseminated encephalomyelitis (ADEM) frequently have LETM, which also occurs in a substantial minority of children with MS and monophasic myelitis. As in adults, antibodies to AQP4 are a critical diagnostic biomarker.17
Coexisting Autoimmunity
Systemic autoimmune disorders such as systemic lupus erythematosus (SLE), antiphospholipid antibody syndrome, and Sjögren’s syndrome often coexist with NMOSD. Clinical episodes concerning for APS, myelitis, or ON in patients with systemic autoimmunity should prompt testing for serum AQP4 antibodies.18 Conversely, patients with NMOSD, especially if seronegative, should be asked about systemic symptoms and have a rheumatologic workup commensurate with the degree of clinical suspicion for Sjögren’s syndrome, SLE, and related conditions.
Diagnosis
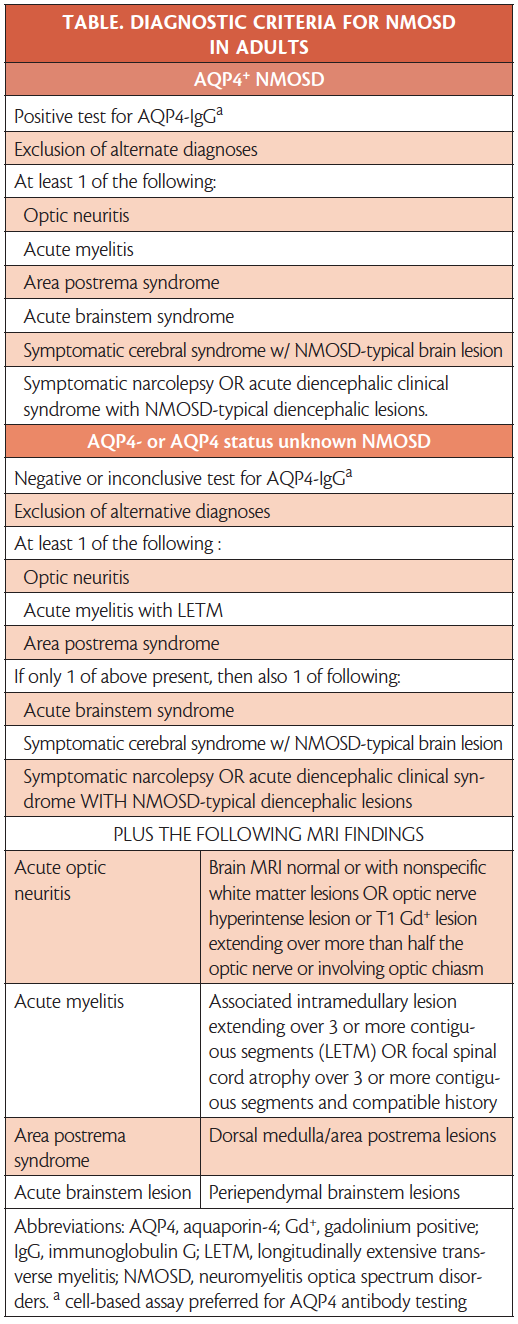
The International Panel for NMO Diagnosis (IPND) updated diagnostic criteria for NMOSD are shown in the Table.10
Laboratory Studies
Serum samples rather than cerebrospinal fluid (CSF) should be used to test for AQP4 antibodies and cell-based assays (eg, fluorescence-activated cell sorting or direct immunofluorescence) should be used.10 Although testing is more sensitive when done prior to initiating immunotherapy, testing should not delay acute treatment. Because even cell-based assays have sensitivity of only 75% to 80%,19 it is prudent to repeat testing after a few weeks or months if results were negative and clinical suspicion is high. Patients with suspected NMOSD should also be tested for serum antibodies to MOG.
Analysis of CSF from patients with NMOSD shows normal to highly inflammatory profiles, the latter being more typical during an episode of myelitis than ON. Pleocytosis (>50 leukocytes/mcL), especially with neutrophilic or eosinophilic predominance, is useful for distinguishing NMOSD from MS, although, most often, both conditions have a lymphocytic predominance. Oligoclonal bands (OCBs) are found in approximately 30% of patients with NMOSD, most often during an attack. In contrast, OCBs are present in more than 85% of patients with MS and pleocytosis of more than 50 leukocytes per mcL is rare in patients with MS.20
Neuroimaging
Neuroimaging features are useful for distinguishing among NMOSD, MS and MOG antibody-associated diseases (Figure 1).21
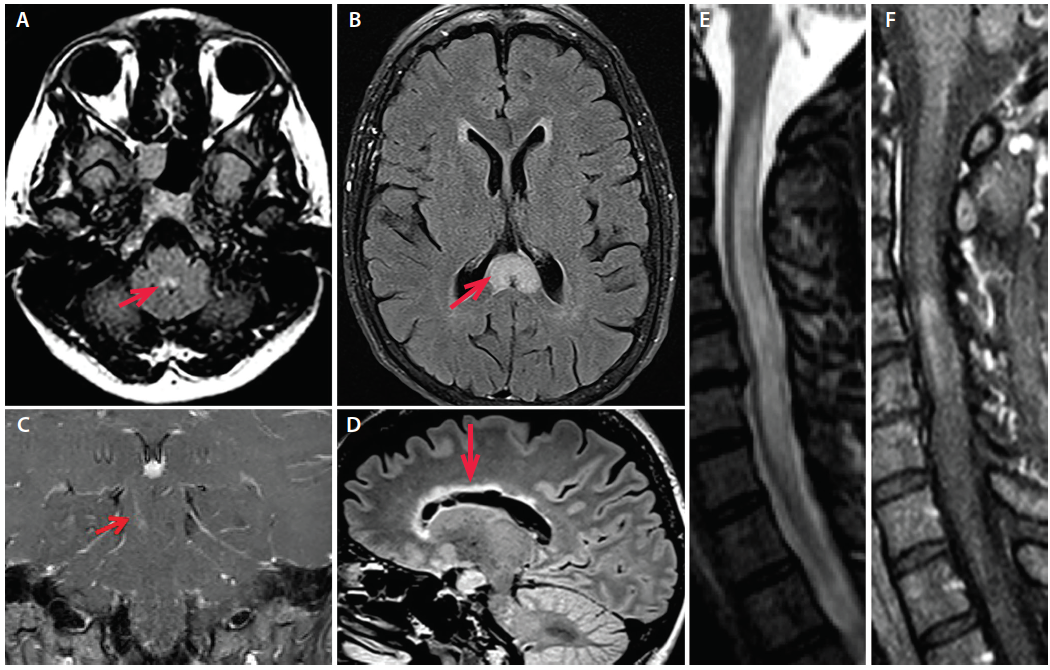
Figure 1. A: Axial fluid-attenuated inversion recovery (FLAIR) MRI of a patient with AQP4+ NMOSD and area postrema syndrome demonstrating T2 hyperintensity in the dorsal medulla (image courtesy of Dr. Divyanshu Dubey). B: Axial FLAIR MRI from a patient with AQP4+ NMOSD demonstrating an “arch bridge” lesion in the splenium of the corpus callosum (arrow; Image courtesy of Dr. Carlos Sollero). C: Coronal T1 postgadolinium MRI in a patient with AQP4+ NMOSD demonstrating an enhancing linear brainstem lesion in the right cerebral peduncle. D: Sagittal FLAIR MRI from a patient with AQP4+ NMOSD demonstrating a marbled lesion in the corpus callosum following the ependymal lining (arrow). E: Cervical spine sagittal STIR MRI of a patient with AQP4+ NMOSD with longitudinally extensive myelitis including F: an area of contrast enhancement on T1 MRI post-gadolinium.
MRI. Inflammation involving more than half the length of the optic nerve, simultaneous bilateral ON, or involvement of the optic chiasm suggest NMOSD, and lesion length correlates with visual prognosis.22 Myelitis in NMOSD tends to involve central gray matter nonexclusively. Although LETM is a hallmark manifestation of NMOSD, it has a broad differential diagnosis including but not limited to infection, neurosarcoidosis, and SLE. Neurosarcoidosis, for example, can produce LETM distinguishable from NMOSD by the presence of constitutional symptoms, pulmonary or hilar adenopathy, hypoglycorrhachia, elevated angiotensin converting enzyme levels, and persistent subpial contrast enhancement.
Corpus callosum lesions occur in 12% to 40% of patients with NMOSD and typically follow the ependymal lining; acute lesions are often edematous and heterogeneous, creating a marbled pattern. The complete thickness of the splenium may be involved, termed the arch bridge pattern. Extensive hemispheric white matter lesions may have a tumefactive appearance resembling Baló’s concentric sclerosis, ADEM or posterior reversible encephalopathy syndrome (PRES). Such large or confluent lesions are more common in children. Lesions surrounding the third ventricle and cerebral aqueduct (including the hypothalamus, ventral midbrain and near the circumventricular organs in the dorsal medulla) are typical of NMOSD. With gadolinium, lesions may appear poorly marginated, subtle, and patchy, termed a cloud-like enhancement pattern. Pencil-thin enhancement of lateral ventricle ependymal surface has been described, as have well-marginated nodular or meningeal areas of enhancement.17
Most lesions seen on MRI in patients with NMOSD are not typical of MS, and only 10% to 20% of patients who have brain lesions will meet Barkhof criteria for MS.17 In common practice across multiple centers, classic MS brain lesions (eg, Dawson’s fingers or subcortical u-fiber lesions) appear useful in distinguishing MS from NMOSD and MOG antibody-associated disease.23,24 The sensitivity and specificity of lesions for distinguishing between MS and NMOSD at disease onset was 79.8% and 87.5%, respectively in a study.24
Optical Coherence Tomography. Widely used to characterize structural injury to the retina in demyelinating diseases (particularly MS), optical coherence tomography (OCT) has shown that ON episodes in patients with NMOSD cause a greater degree of retinal damage than in patients with MS (Figure 2).25 Studies with OCT have also demonstrated that retinal damage in NMOSD can occur independently of ON attacks, raising the possibility that neurodegeneration may contribute to disability, in addition to the relapse-driven disability most commonly attributed to AQP4+ NMOSD.25,26
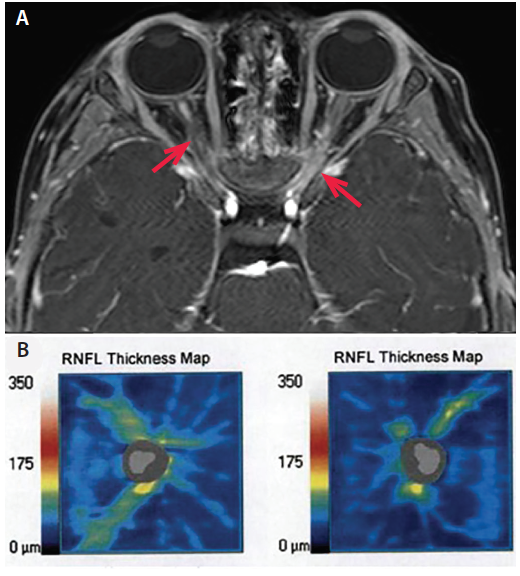
Figure 2. A: Axial T1 fat-saturated post-gadolinium MRI in a patient with AQP4+ NMOSD demonstrates bilateral optic neuritis over more than 50% of the optic nerves (arrows). B: Optical coherence tomography taken 1 year after bilateral optic neuritis demonstrates thinning of the right and left retinal nerve fiber layers (right 62 μm, left 54 μm; normal thickness on OCT is more than 80 μm; OCT images contributed by Dr. Sara Qureshi).
Treatment
Treatment of Acute Relapses
Because most disability in NMOSD accumulates as a direct consequence of relapses that typically leave substantial residual disability, a relapse can be considered an emergency, calling for prompt neurologic examination and evaluation for occult infection to identify pseudorelapses. Vision loss may be the only early predictor of true relapse.27 Emergent spinal MRI may be necessary for patients with new or changing symptoms and a prior episode of myelitis, particularly when signs and symptoms are not clearly localizable to a new lesion. High clinical suspicion, aggressive acute treatment, and prompt initiation of preventative immunotherapy are critical.
Given our understanding of the pathophysiology of NMOSD, our practice is to administer intravenous methylprednisolone (IVMP) and plasma exchange (PLEX) as first-line acute therapy as quickly as possible for most patients with NMOSD when the clinical presentation is consistent with an exacerbation, especially those with AQP4+ NMOSD.
Corticosteroids. Acute exacerbations of NMOSD are initially treated with IVMP, typically 1 g daily for 3 to 5 days. Some clinicians include an oral prednisone taper starting at up to 1 mg per kg per day (usually 15-30 mg/day) and decreasing gradually over several weeks depending on individual factors such as timing of disease modifying therapy, patient comorbidities, and tolerance of oral glucocorticoids. Long-term oral glucocorticoids are associated with significant toxicity and are often avoided in favor of steroid-sparing immunotherapy.
Apheresis therapy (plasma exchange or immunoadsorption). Because antibodies to AQP4 activate the complement cascade leading to demyelination and necrosis with loss of astrocytes, axons, and oligodendrocytes, many clinicians, including the authors, advocate for early PLEX rather than using it only as a rescue therapy for patients who are not improving with IVMP. A retrospective study demonstrated improved outcomes when patients were treated with PLEX early in the course of an exacerbation.28 Probability of complete recovery decreased from 50% with PLEX immediately (day 0), to 1% to 5% after day 20, emphasizing the importance of early treatment. In another study, 51% of patients treated with IVMP and PLEX at day 5 after presentation recovered to prerelapse Expanded Disability Status Scale (EDSS) baseline compared with only 16.6% of patients who received IVMP only.29 Retrospective analysis of 871 patients having NMOSD attacks found the addition of PLEX to IVMP improved responder rates and that first-line PLEX was superior to IVMP for myelitis.30 Immediate initiation of apheresis (within 2 days of symptom onset) was associated with a 40% rate of complete response compared with 3.2% when started at 6 days after symptom onset, again emphasizing the importance of early PLEX.31 Strong predictors of a complete response were the use of apheresis as first-line therapy, time from onset of attack to start of therapy in days, presence of antibodies to AQP4, and monofocal attack. There was no significant difference between PLEX and immunoadsorption.
Intravenous Immunoglobulins. Data on the use of intravenous immunoglobulin (IVIG) in patients with NMO relapses remain limited, although small studies suggest that IVIG may be effective in preventing relapses.32,33
Relapse Prevention
We recommend prompt initiation of long-term immunosuppression once a diagnosis of NMOSD is made.34 No randomized clinical trial investigating preventative therapy in NMOSD has had final results published to date, therefore treatment is based on open-label prospective and retrospective studies. Given the available data, we prefer rituximab (RTX) for most patients with NMOSD. If there are contraindications to RTX, we typically use mycophenolate mofetil (MMF).
To date, the best-studied immunotherapies for NMOSD include RTX, MMF, and azathioprine (AZA). Treatment with RTX or MMF, and to a lesser degree, AZA, is associated with a decrease in relapse rates in patients with NMOSD; retrospective studies suggest that relapse risk is significantly higher with AZA compared with MMF or RTX.35,36 Treatment with RTX decreased relapse rate by up to 88%, with 2 of 3 patients achieving complete remission during a 10-year study period, whereas MMF reduced relapse rates by 87.4% with a 36% failure rate and AZA reduced relapse rates by 72% with a 53% failure rate even when used concurrently with prednisone.35 A retrospective review of 116 patients found predictors of poor response to AZA or MMF included pretreatment history of severe attack and younger age. Among 40 patients with a poor response to oral agents, 29 were changed to RTX, and among those, only 10% had a poor response to RTX.37
Adequate RTX dose is important in order to maintain remission. Although the optimal dosing regimen has yet to be determined, we agree with dosing protocols tailored to an individual’s rate of immune reconstitution. A target B-cell (CD19+ and CD20+) count less than 0.05% of total lymphocytes in the first 2 years and less than 0.1% thereafter can be considered.38 Some practices are monitoring the number of CD27+ cells in isolation as there is evidence that these are a marker for the return of disease activity.39 Because RTX can produce hypogammaglobulinemia which may be associated with significant infections, we monitor immunoglobulin levels prior to initiation and maintenance doses of RTX.40 Patients must also be counseled regarding the risk of infusion reactions and infections. No studies of ocrelizumab (an anti-CD20 monoclonal antibody approved for treating relapsing and primary progressive MS) for NMOSD have been reported.
Breakthrough Disease
If a relapse is confirmed while on immunotherapy, it is important to determine if the medication is being optimally dosed. For RTX, B-cell reconstitution should be investigated and dosing adjusted to optimize suppression of memory B cells. Based on clinical experience, we consider PLEX just prior to RTX infusions for patients with appropriately suppressed memory B cells who have a breakthrough relapse. Ongoing clinical trials will likely provide additional options for such patients. For patients on MMF, the lymphocyte count can be targeted to less than 1,500 per mcL and dose increased up to the maximum dose of 3 g per day if necessary.
Treatment During Pregnancy
Risks of withholding acute treatment, including irreversible paralysis and blindness for the mother must be weighed carefully against risks of treating relapses during pregnancy. Lower infant head circumference and birth weight have been reported with corticosteroid exposure during pregnancy. Although historical data suggested corticosteroid exposure increased risks of cleft lip or palate, more recent studies have not reproduced this and suggest that the risk, if any, is very small.41 Plasma exchange has been administered safely for pregnant women with NMOSD relapse at various gestational stages.15
For preventative therapy, methotrexate and cyclophosphamide are contraindicated during pregnancy, whereas AZA and RTX appear to have better safety profiles.15 Although listed by the Food and Drug Administration (FDA) as pregnancy class D, in 3,124 women exposed to AZA during pregnancy, there was no increased risk of preterm birth, low birth weight, miscarriage, birth defects, major congenital malformations or neoplasia compared with that seen in unexposed mothers.15 Although RTX crosses the placenta after gestational weeks 16 to 20 and depletes fetal B cells, miscarriage and congenital malformation rates in children exposed prenatally are similar to that seen in the general population and B-cell depletion was associated with serious infection in only 1 of 153 offspring in a study.42 Given the relative safety of RTX, patients can be treated with RTX well before planned conception (eg, a few months) and again in the early postpartum period. Monthly IVIG has also been used, although there is less data regarding efficacy.32,33
Treatment in Children
Treatment of pediatric NMOSD is largely similar to treatment in adults,43 although pediatric patients treated with RTX often require more frequent dosing as their B cells tend to reconstitute more rapidly than in adults.
Medications to Avoid in NMOSD
Several studies have suggested inefficacy or worsening of NMOSD associated with the use of some disease modifying agents used to treat MS, including interferon β,44 natalizumab,45 and fingolimod.46 Therefore, we avoid these agents for NMOSD.
Future Directions
Since 2014, clinical trials for several agents with potential to treat patients with NMOSD have been launched, including tocilizumab, satralizumab, eculizumab, and inebilizumab.47
Tocilizumab
Two open-label trials of tocilizumab, an interleukin-6 (IL-6) receptor antagonist, demonstrated a reduction in relapse rates in patients with AQP4+ NMOSD as adjunctive treatment or monotherapy.48,49 In one of these, 7 patients had an ARR reduction from 2.9 (despite background immunotherapy, including AZA or corticosteroids) to 0.4 with the addition of tocilizumab.49 In the other, 8 patients who were refractory to RTX treatment had an ARR reduction from 4.0 to 0.4 after treatment with tocilizumab.48 There was significant reduction in neuropathic pain for patients who received tocilizumab in both studies.
Satralizumab
Based on the success of tocilizumab, which requires dosing every 1-2 weeks, satralizumab (SA237) was developed. It is a humanized recycling monoclonal antibody targeting the IL-6 receptor with extended dosing frequency of once per month or longer. Preliminary results from a randomized, international, double blind phase 3 trial of satralizumab as adjunctive therapy showed satralizumab reduced the risk of relapse by 62% in patients with NMOSD (n = 83); those who were AQP4+ (n = 53) had a 79% risk reduction and who were AQP4- (n = 30) had a 34% risk reduction.50
Eculizumab
Eculizumab is a monoclonal antibody that inhibits activation of C5 complement protein, thereby disrupting the complement cascade. In an open-label pilot study, 12 of 14 patients with AQP4+ NMOSD (6 who had failed prior immunotherapy) had no attacks and 2 had possible minor single attacks while being treated with eculizumab for 48 weeks. After the trial, when returned to alternative immunotherapy (AZA, RTX, MMF, or prednisone), 5 had a relapse in the subsequent year. Hemolytic complement activity dropped from a mean of 88.5% to 0.4% after 6 weeks of treatment and remained suppressed during the study.51
An open-label phase 3 double-blind placebo-controlled randomized trial of eculizumab in patients with AQP4+ NMOSD that permits patients to remain on certain background immunosuppressants to attenuate the placebo risk is ongoing. Eculizumab shows great promise, including in treatment-resistant NMOSD. However, use is associated with increased risk of infection by encapsulated bacteria that includes more than a 1,000-fold increased risk of meningococcal disease.52
Inebilizumab
Inebilizumab (MEDI551) is a humanized monoclonal antibody against CD19 that targets B cells, B-cell precursors, and plasmablasts. The rationale for the considering inebilizumab is based on observations with RTX. Inebilizumab is currently being studied in a phase 3 clinical trial during which patients are randomized in a 3:1 ratio (inebilizumab to placebo) and those assigned placebo are transitioned to an open-label phase after 6.5 months. In results from the N-MOmentuma trial announced via press release, inebilizumab treatment of patients with NMOSD reduced the risk of relapse by 77% compared with treatment with placebo. Primary and secondary endpoints of the trial were reportedly achieved.
Other Investigational Drugs
An open-label phase 1b trial of a C1-esterase inhibitor added to IVMP for patients with NMO/NMOSD relapses demonstrated safety and preliminary evidence for improved outcomes.53 Bortezomib is a proteasome inhibitor that depletes plasma cells and was given as adjunctive therapy to 5 patients with AQP4+ NMOSD who were not responsive to standard therapy. Clinical stability was seen in 5 of 5 patients and 80% of patients achieve relapse-free status.54 In an open-label prospective pilot study of 16 patients with AQP4+ NMOSD, oral cetirizine (10 mg) daily added to current immunotherapy was well-tolerated and associated with a marginal but significant reduction in ARR.55
Conclusions
The diagnostic criteria for NMOSD were updated in 2015 and account for both patients who are AQP4 seropositive and seronegative. Treatment recommendations have been largely based on retrospective studies and expert clinical experience with data supporting RTX, MMF, or AZA. Ongoing clinical trials may offer more evidence for using new therapies in the near future. It is critical that neurologists encountering patients with suspected demyelinating disorders consider NMOSD in the differential diagnosis, thereby avoiding delays in recognition and treatment.
a. A double-masked, placebo-controlled study with open label period to evaluate MEDI-551 in neuromyelitis optica and neuromyelitis optica spectrum disorders (NCT02200770).
1. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106-2112.
2. Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm. 2015;2:e110.
3. Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017;264:2088-2094.
4. Flanagan EP, Cabre P, Weinshenker BG, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol. 2016;79:775-783.
5. Etemadifar M, Nasr Z, Khalili B, Taherioun M, Vosoughi R. Epidemiology of neuromyelitis optica in the world: a systematic review and meta-analysis. Mult Scler Int. 2015;2015:174720.
6. Viegas S, Weir A, Esiri M, et al. Symptomatic, radiological and pathological involvement of the hypothalamus in neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2009;80:679-682.
7. Nakajima H, Hosokawa T, Sugino M, et al. Visual field defects of optic neuritis in neuromyelitis optica compared with multiple sclerosis. BMC Neurol. 2010;10:45.
8. Lim YM, Pyun SY, Lim HT, Jeong IH, Kim KK. First-ever optic neuritis: distinguishing subsequent neuromyelitis optica from multiple sclerosis. Neurol Sci. 2014;35:781-783.
9. Popescu BF, Lennon VA, Parisi JE, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology. 2011;76:1229-1237.
10. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177-189.
11. Shosha E, Dubey D, Palace J, et al. Area postrema syndrome: frequency, criteria, and severity in AQP4-IgG-positive NMOSD. Neurology. 2018;91(17):e1642-e1651.
12. Kremer L, Mealy M, Jacob A, et al. Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Mult Scler. 2014;20:843-847.
13. Flanagan EP, Weinshenker BG, Krecke KN, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol. 2015;72:81-87.
14. Houtchens MK, Edwards NC, Phillips AL. Relapses and disease-modifying drug treatment in pregnancy and live birth in US women with MS. Neurology. 2018;91:e1570-e1578.
15. Shosha E, Pittock SJ, Flanagan E, Weinshenker BG. Neuromyelitis optica spectrum disorders and pregnancy: interactions and management. Mult Scler. 2017;23:1808-1817.
16. Bradshaw MJ, Vu N, Hunley TE, Chitnis T. Child neurology: neuromyelitis optica spectrum disorders. Neurology. 2017;88:e10-e13.
17. Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. 2015;84:1165-1173.
18. Wingerchuk DM, Weinshenker BG. The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler. 2012;18:5-10.
19. Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78:665-671; discussion 669.
20. Dobson R, Ramagopalan S, Davis A, Giovannoni G. Cerebrospinal fluid oligoclonal bands in multiple sclerosis and clinically isolated syndromes: a meta-analysis of prevalence, prognosis and effect of latitude. J Neurol Neurosurg Psychiatry. 2013;84:909-914.
21. Bensi C, Marrodan M, Gonzalez A, et al. Brain and spinal cord lesion criteria distinguishes AQP4-positive neuromyelitis optica and MOG-positive disease from multiple sclerosis. Mult Scler Relat Disord. 2018;25:246-250.
22. Akaishi T, Nakashima I, Takeshita T, et al. Lesion length of optic neuritis impacts visual prognosis in neuromyelitis optica. J Neuroimmunol. 2016;293:28-33.
23. Jurynczyk M, Tackley G, Kong Y, et al. Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry. 2017;88:132-136.
24. Hyun JW, Huh SY, Shin HJ, et al. Evaluation of brain lesion distribution criteria at disease onset in differentiating MS from NMOSD and MOG-IgG-associated encephalomyelitis. Mult Scler. 2018:1352458518761186.
25. Peng A, Qiu X, Zhang L, et al. Evaluation of the retinal nerve fiber layer in neuromyelitis optica spectrum disorders: a systematic review and meta-analysis. J. Neurol Sci. 2017;383:108-113.
26. Oertel FC, Havla J, Roca-Fernandez A, et al. Retinal ganglion cell loss in neuromyelitis optica: a longitudinal study. J Neurol Neurosurg Psychiatry. 2018;89:1259-1265.
27. Kessler RA, Mealy MA, Levy M. Early indicators of relapses vs pseudorelapses in neuromyelitis optica spectrum disorder. Neurol Neuroimmnol Neuroinflamm. 2016;3:e269.
28. Bonnan M, Valentino R, Debeugny S, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89:346-351.
29. Abboud H, Petrak A, Mealy M, Sasidharan S, Siddique L, Levy M. Treatment of acute relapses in neuromyelitis optica: steroids alone versus steroids plus plasma exchange. Mult Scler. 2016;22:185-192.
30. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79:206-216.
31. Kleiter I, Gahlen A, Borisow N, et al. Apheresis therapies for NMOSD attacks: A retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm. 2018;5:e504.
32. Elsone L, Panicker J, Mutch K, Boggild M, Appleton R, Jacob A. Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: experience in 10 patients. Mult Scler. 2014;20:501-504.
33. Viswanathan S, Wong AH, Quek AM, Yuki N. Intravenous immunoglobulin may reduce relapse frequency in neuromyelitis optica. J Neuroimmunol. 2015;282:92-96.
34. Kimbrough DJ, Fujihara K, Jacob A, et al. Treatment of neuromyelitis optica: review and recommendations. Mult Scler Relat Disord. 2012;1:180-187.
35. Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol. 2014;71:324-330.
36. Qiu W, Kermode AG, Li R, et al. Azathioprine plus corticosteroid treatment in Chinese patients with neuromyelitis optica. J Clin Neurosci. 2015;22:1178-1182.
37. Kim SH, Hyun JW, Joung A, Park EY, Joo J, Kim HJ. Predictors of response to first-line immunosuppressive therapy in neuromyelitis optica spectrum disorders. Mult Scler. 2017;23:1902-1908.
38. Kim SH, Hyun JW, Kim HJ. Individualized B cell-targeting therapy for neuromyelitis optica spectrum disorder. Neurochem Int. 2018.
39. Restoration of regulatory B cell deficiency following alemtuzumab therapy in patients with relapsing multiple sclerosis. J Neuroinflamm. 2018;15:300.
40. Marcinno A, Marnetto F, Valentino P, et al. Rituximab-induced hypogammaglobulinemia in patients with neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm. 2018;5:e498.
41. Xiao WL, Liu XY, Liu YS, Zhang DZ, Xue LF. The relationship between maternal corticosteroid use and orofacial clefts-a meta-analysis. Reprod Toxicol. 2017;69:99-105.
42. Chakravarty EF, Murray ER, Kelman A, Farmer P. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
43. Gombolay GY, Chitnis T. Pediatric neuromyelitis optica spectrum disorders. Curr Treat Options Neurol. 2018;20:19.
44. Shimizu J, Hatanaka Y, Hasegawa M, et al. IFNbeta-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology. 2010;75:1423-1427.
45. Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79:1065-1066.
46. Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler. 2012;18:113-115.
47. Paul F, Murphy O, Pardo S, Levy M. Investigational drugs in development to prevent neuromyelitis optica relapses. Exp Opin Invest Drugs. 2018;27:265-271.
48. Ringelstein M, Ayzenberg I, Harmel J, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015;72:756-763.
49. Araki M, Matsuoka T, Miyamoto K, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology. 2014;82:1302-1306.
50. Yamamura T, Kleiter I, Fujihara K, et al. A double-blind placebo-controlled study of satralizumab (SA237), a recycling anti-IL-6 receptor monoclonal antibody, as add-on therapy for neuromyelitis optica spectrum disorder (NMOSD). Presented at: European Congress for Treatment and Research in Multiple Sclerosis; October, 2018: Abstract 536.
51. Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurology 2013;12:554-562.
52. McNamara LA, Topaz N, Wang X, et al. High risk for invasive meningococcal disease among patients receiving eculizumab (soliris) despite receipt of meningococcal vaccine. Am J Transplant. 2017;17:2481-2484.
53. Levy M, Mealy MA. Purified human C1-esterase inhibitor is safe in acute relapses of neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. 2014;1:e5.
54. Zhang C, Tian DC, Yang CS, et al. Safety and efficacy of bortezomib in patients with highly relapsing neuromyelitis optica spectrum disorder. JAMA Neurol. 2017;74:1010-1012.
55. Sand IK, Telford R, Kraus TA, et al. Open-label, add-on trial of cetirizine for neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. 2018;5(2):e441.
Disclosures
MJB serves on the editorial board of Continuum: Lifelong Learning in Neurology. The authors have no financial relationships relevant to this content to disclose.


